17 2 June 2017
17.1 Patterns of silique variation, continued
To get around the confusing results last time, let’s just create a new factor variable that is Gen:ID, so that each combo is treated independently.
sil_data <- subset(popLer_sil, Treatment == "B" & Generation > 0 & Gap == "0p")
sil_data$GenID <- with(sil_data, interaction(Gen, ID))
summary(aov(Siliques ~ Gen + GenID, data = sil_data)) Df Sum Sq Mean Sq F value Pr(>F)
Gen 4 116793 29198 16.38 4.22e-12 ***
GenID 47 266454 5669 3.18 1.28e-09 ***
Residuals 288 513482 1783
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1OK, that’s better clarity. After accounting for inter-generational variation, most of the remaining variation is among runways rather than within runways.
ggplot(aes(Gen, Siliques, group = ID, color = ID), data = sil_data) + geom_point() +
geom_smooth(aes(Generation, Siliques), span = 1, se = FALSE)`geom_smooth()` using method = 'loess'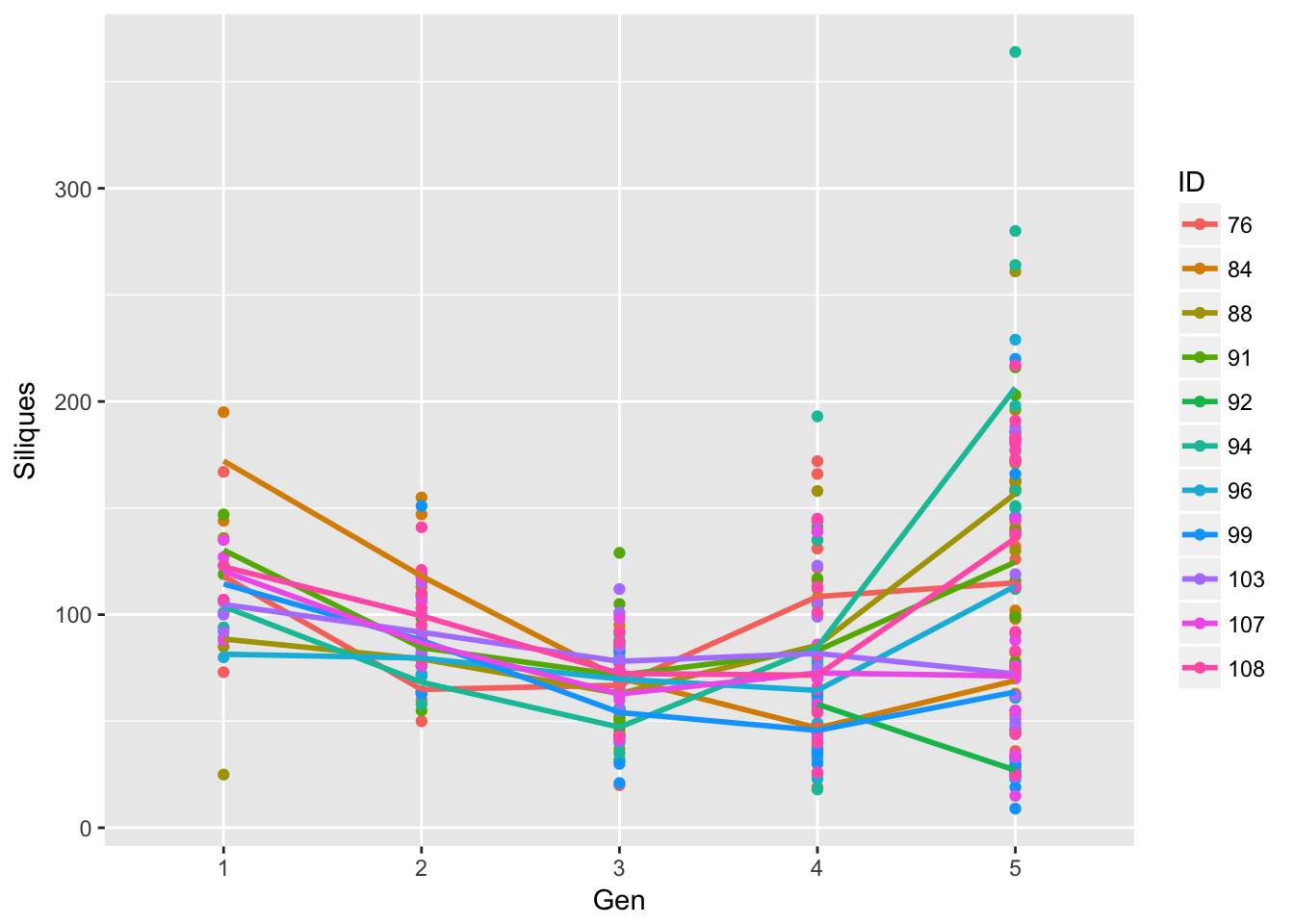
Let’s look at how the residual variance scales with the mean.
sil_stats <- group_by(sil_data, Gen, ID) %>%
summarise(Mean = mean(Siliques),
Var = var(Siliques),
SD = sqrt(Var))
sil_stats <- sil_stats[complete.cases(sil_stats), ]
ggplot(aes(Mean, Var, color = ID), data = sil_stats) + geom_point() 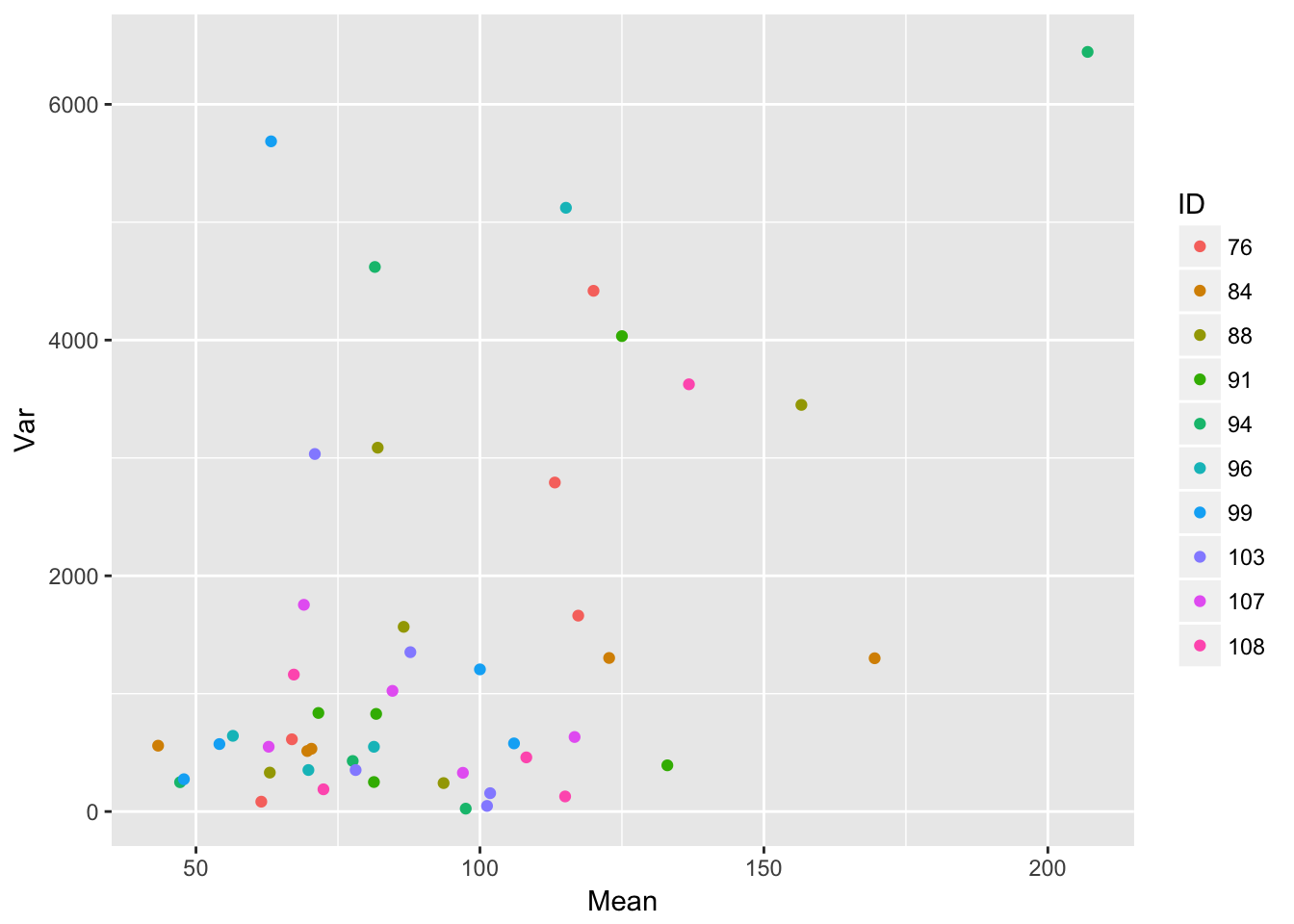
ggplot(aes(Mean, SD, color = ID), data = sil_stats) + geom_point() 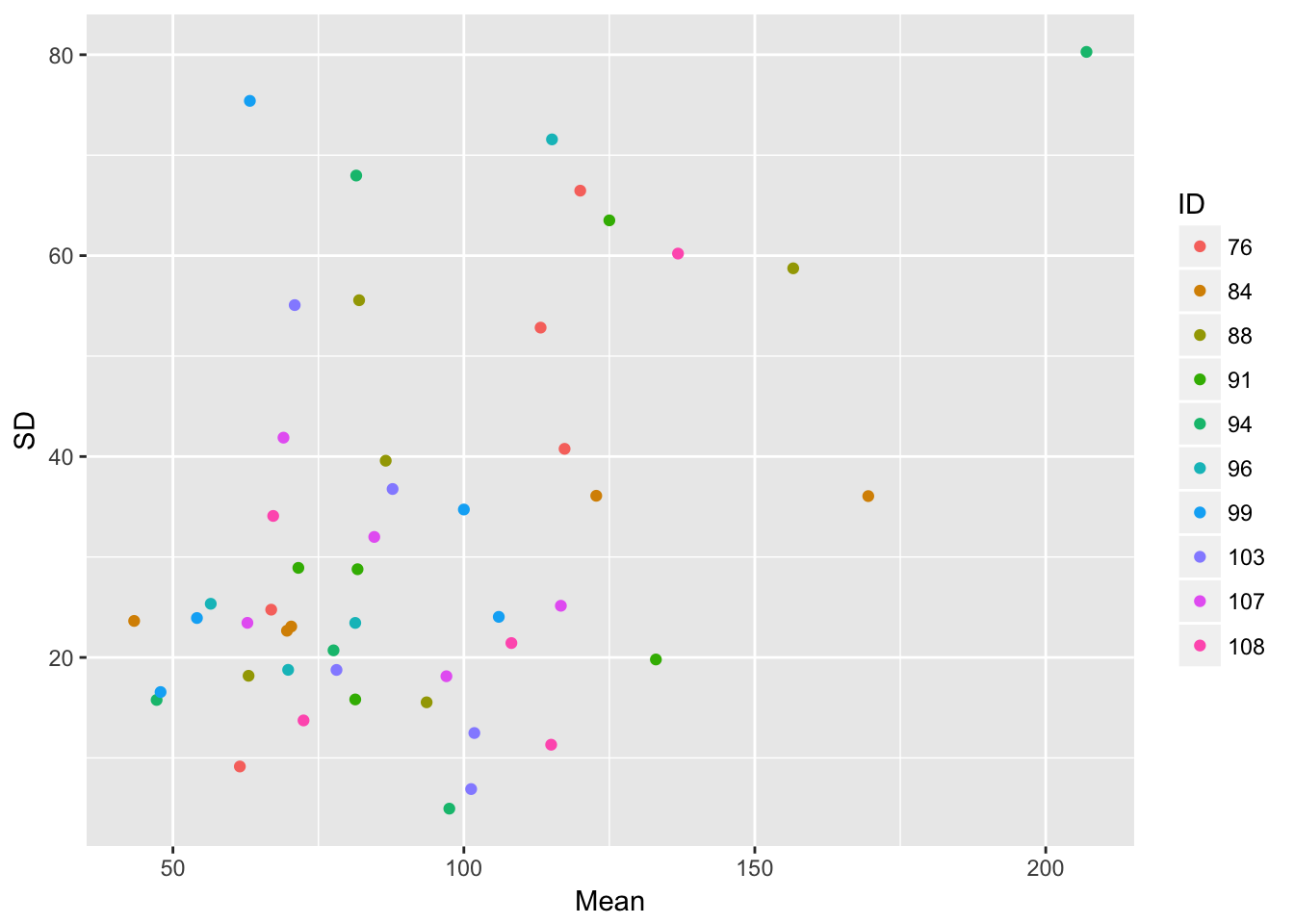
ggplot(aes(Mean, Var), data = sil_stats) + geom_point() + geom_smooth(method = "lm")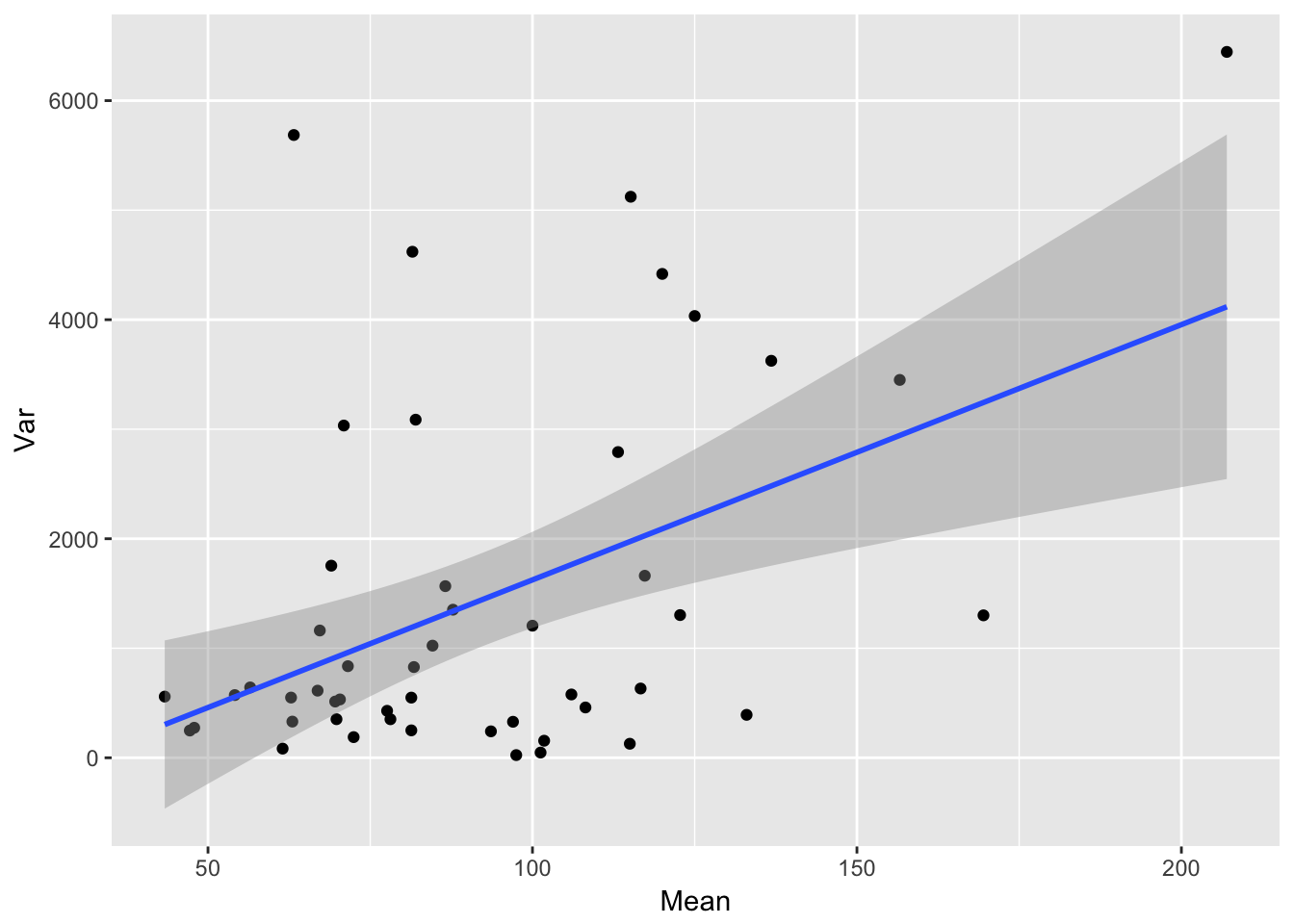
ggplot(aes(Mean, SD), data = sil_stats) + geom_point() + geom_smooth(method = "lm")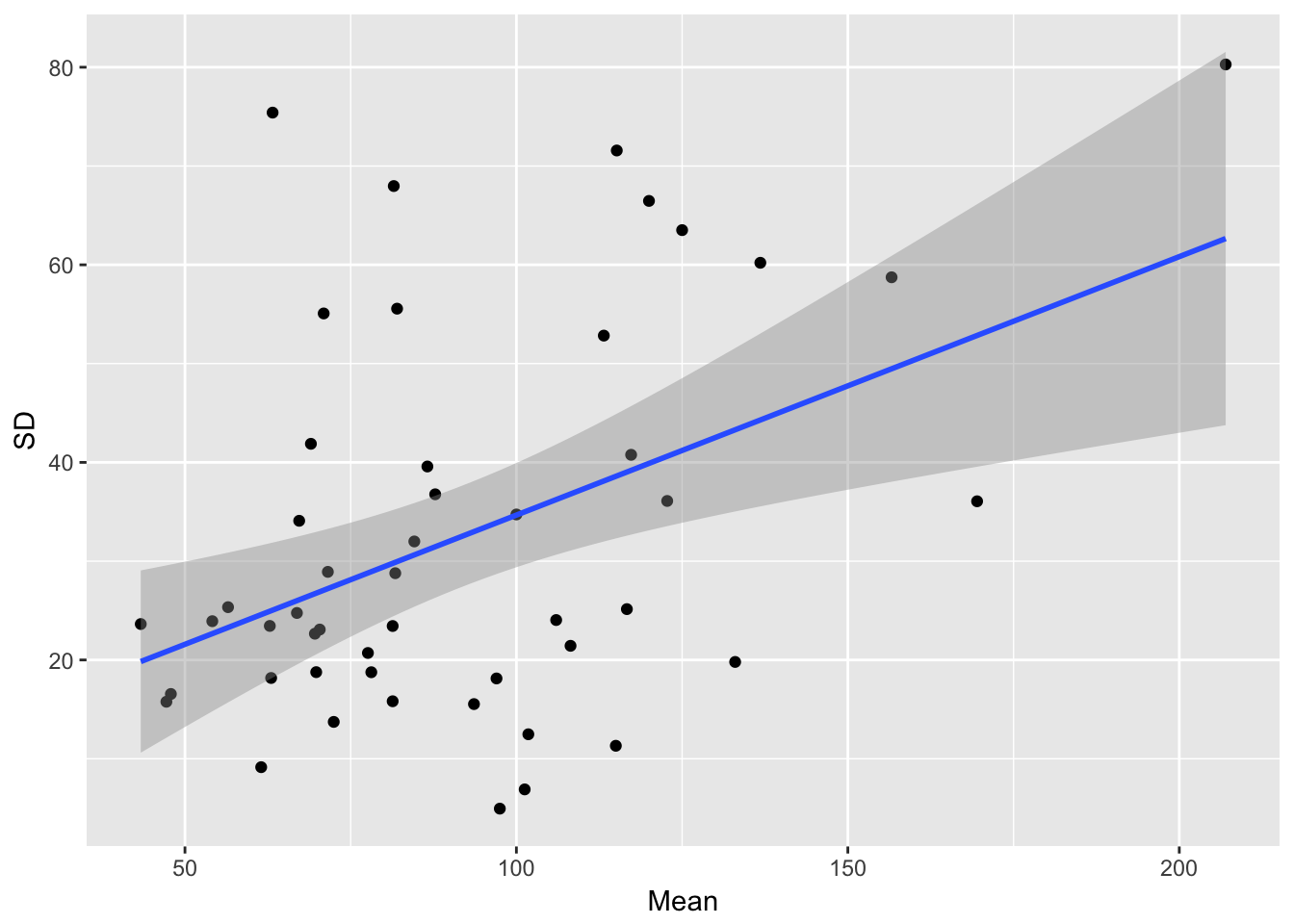
The relationship betwen the SD and mean looks a bit more linear, although there’s still a lot of scatter.
Now let’s see if the seed:silique ratio is structured in any way, or is independent among pots.
sil_data <- subset(popLer_sil, Treatment == "B" & Gap == "3p")
sil_data$Generation <- sil_data$Generation + 1
sil_data$Gen <- as.factor(sil_data$Generation)
popLer$ID <- as.factor(popLer$ID)
sil_seed_data <- left_join(sil_data, popLer)Joining, by = c("Generation", "ID", "Treatment", "Gap", "Pot", "Gen")Warning in left_join_impl(x, y, by$x, by$y, suffix$x, suffix$y): joining
factors with different levels, coercing to character vector
Warning in left_join_impl(x, y, by$x, by$y, suffix$x, suffix$y): joining
factors with different levels, coercing to character vectorWarning in left_join_impl(x, y, by$x, by$y, suffix$x, suffix$y): joining
factor and character vector, coercing into character vectorggplot(aes(Siliques, Seedlings, color = Gen), data = sil_seed_data) +
geom_point() + geom_smooth(method = "lm")Warning: Removed 2 rows containing non-finite values (stat_smooth).Warning: Removed 2 rows containing missing values (geom_point).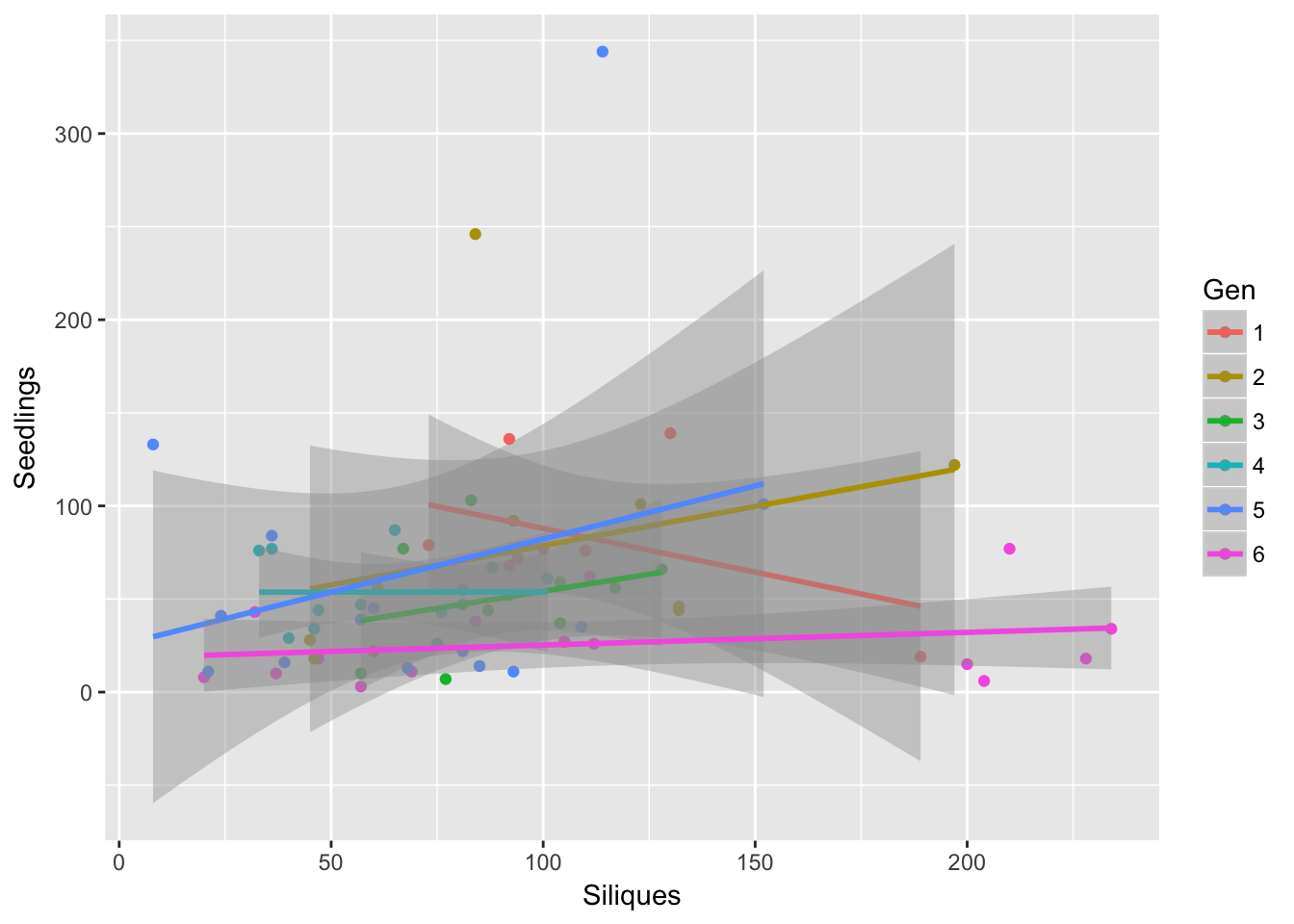
Within each generation, the number of seedlings in the home pot is independent of the number of siliques!!!
Now include the dispersing seeds in gen 1.
sil_data <- subset(popLer_sil, Treatment == "B" & Gap == "0p" & Gen == "0")
sil_data$Generation <- sil_data$Generation + 1
sil_data$Gen <- as.factor(sil_data$Generation)
Ler_seed_gen1$ID <- as.factor(Ler_seed_gen1$ID)
sil_seed_data <- left_join(sil_data, Ler_seed_gen1)Joining, by = "ID"Warning in left_join_impl(x, y, by$x, by$y, suffix$x, suffix$y): joining
factors with different levels, coercing to character vectorggplot(aes(Siliques, eff_sd_no), data = sil_seed_data) + geom_point() +
geom_smooth(method = "lm")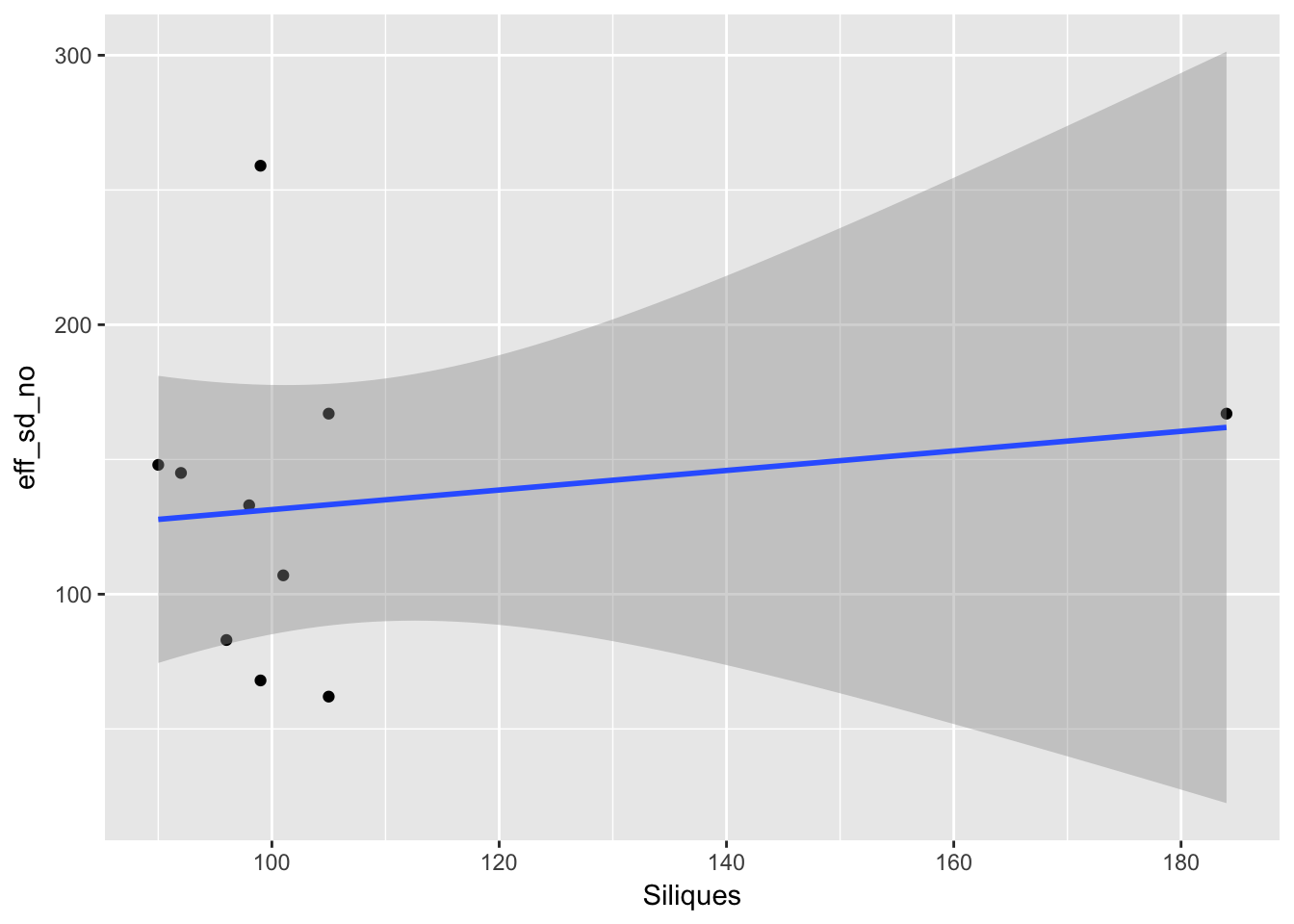
ggplot(aes(Siliques, home), data = sil_seed_data) + geom_point() +
geom_smooth(method = "lm")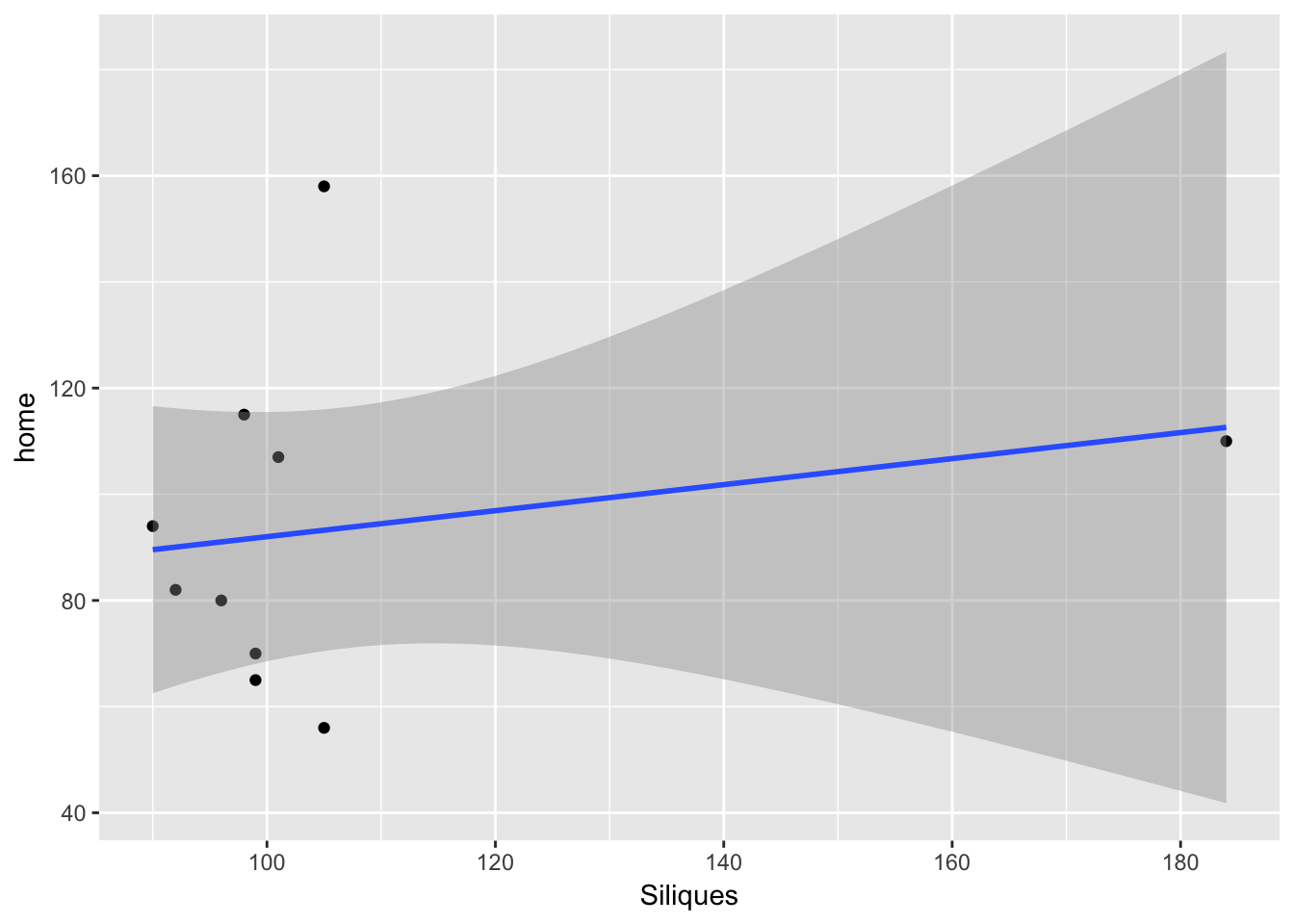
ggplot(aes(Siliques, stay_prop), data = sil_seed_data) + geom_point() +
geom_smooth(method = "lm")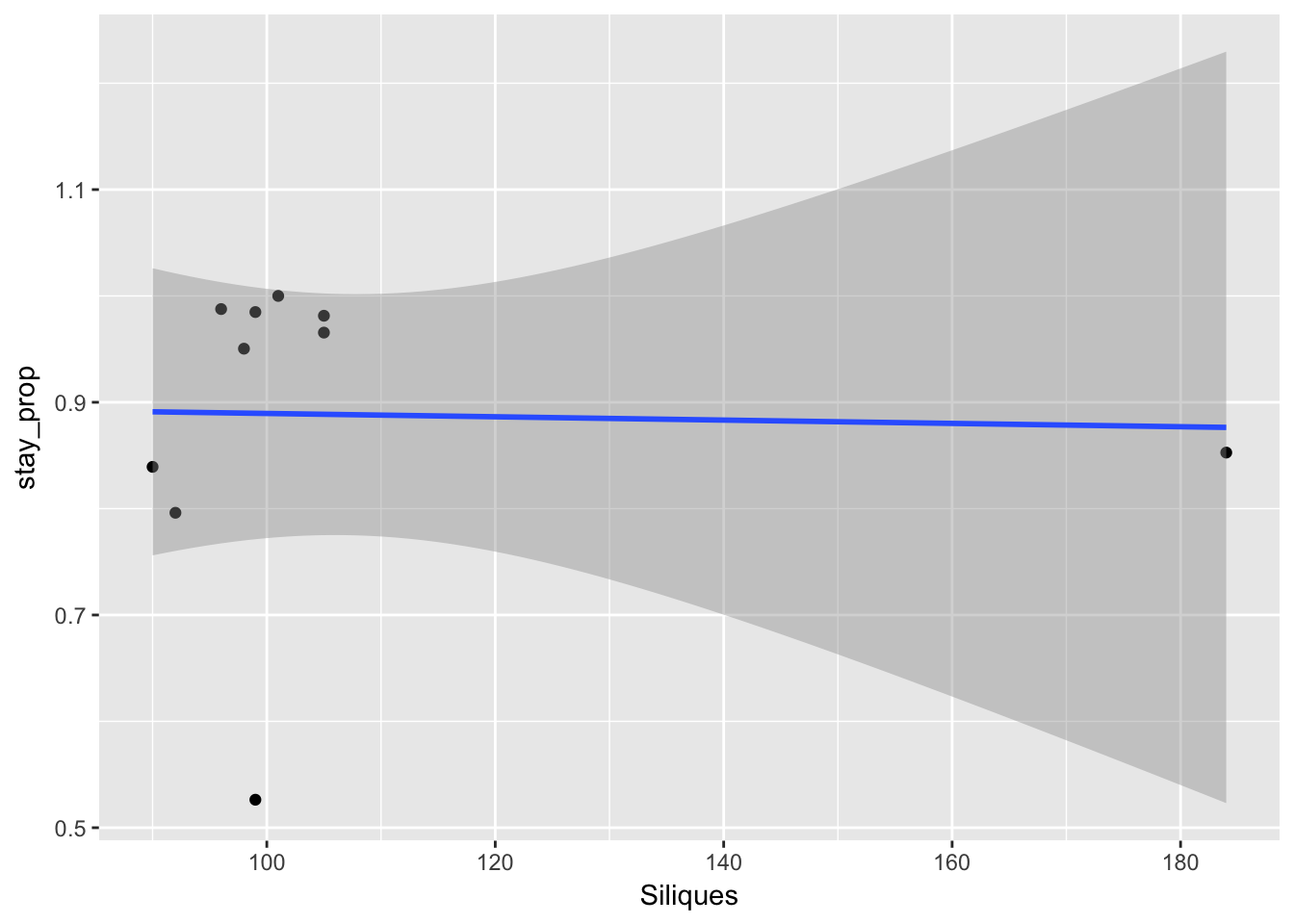
So the lack of relationship between siliques and seedling is not because of variability in dispersal.
Let’s look at population structure in the variability of home pot seedlings, using the 1p gaps to get replication within populations but seedling numbers mostly from local production:
seed_data <- subset(popLer, Treatment == "B" & Generation > 1 & Gap == "1p")
seed_data$GenID <- with(seed_data, interaction(Gen, ID))
summary(aov(Seedlings ~ Gen + GenID, data = seed_data)) Df Sum Sq Mean Sq F value Pr(>F)
Gen 4 31628 7907 5.747 0.000388 ***
GenID 45 178349 3963 2.881 1.41e-05 ***
Residuals 84 115567 1376
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1ggplot(aes(Gen, Seedlings, group = ID, color = ID), data = seed_data) + geom_point() +
geom_smooth(span = 1, se = FALSE)`geom_smooth()` using method = 'loess'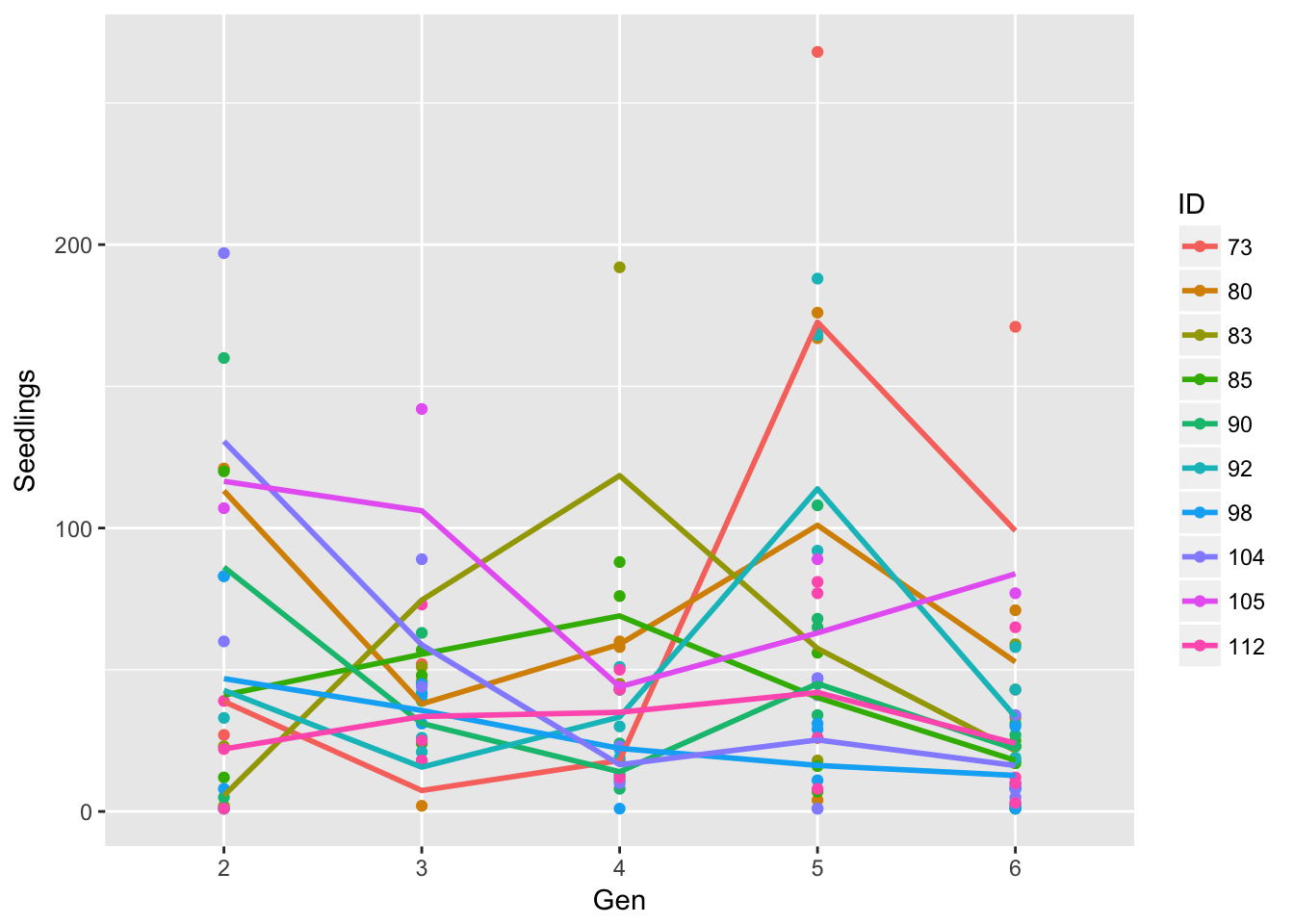
Let’s look at how the residual variance scales with the mean.
sil_stats <- group_by(seed_data, Gen, ID) %>%
summarise(Mean = mean(Seedlings),
Var = var(Seedlings),
SD = sqrt(Var))
sil_stats <- sil_stats[complete.cases(sil_stats), ]
ggplot(aes(Mean, Var, color = ID), data = sil_stats) + geom_point() 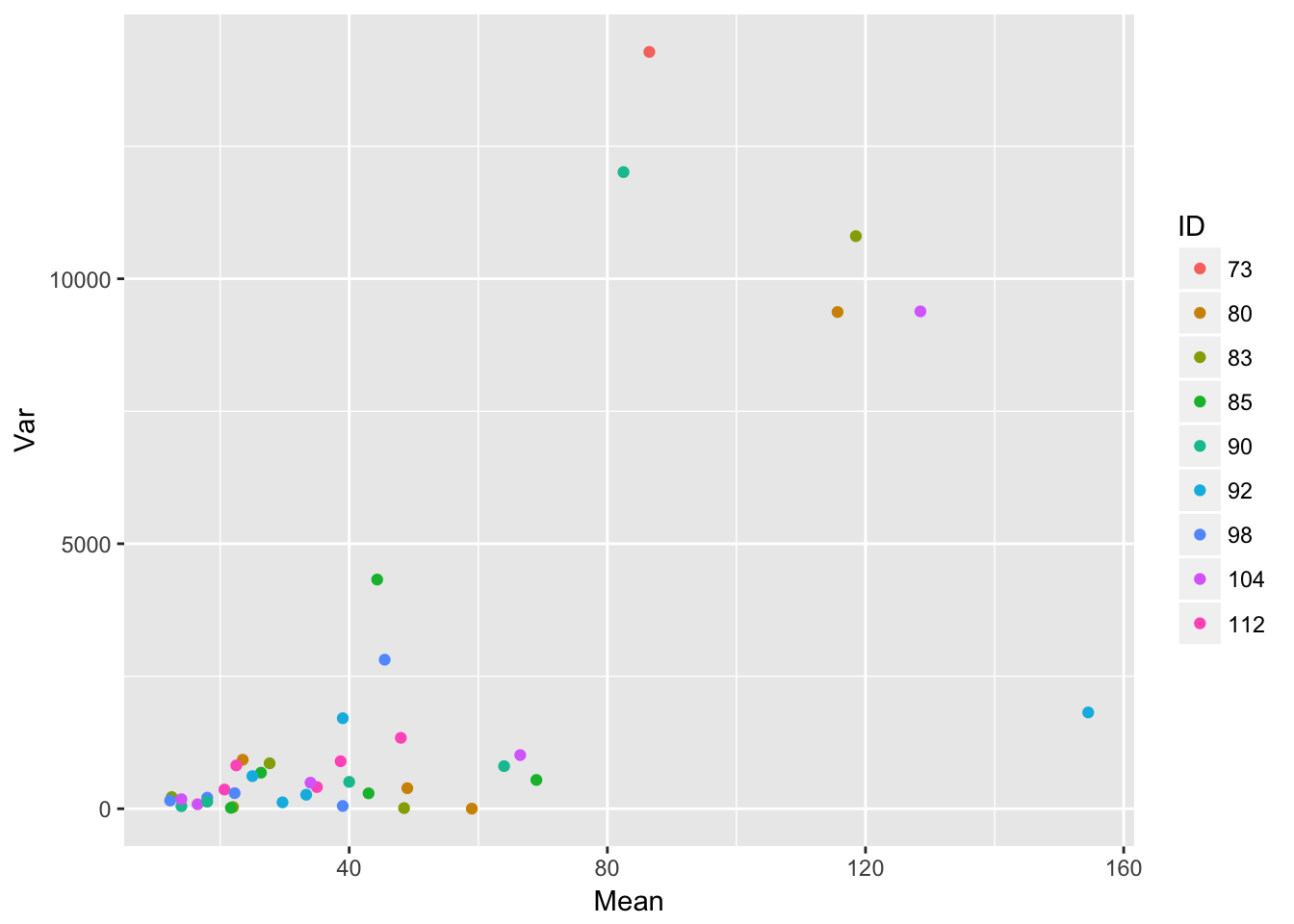
ggplot(aes(Mean, SD, color = ID), data = sil_stats) + geom_point() 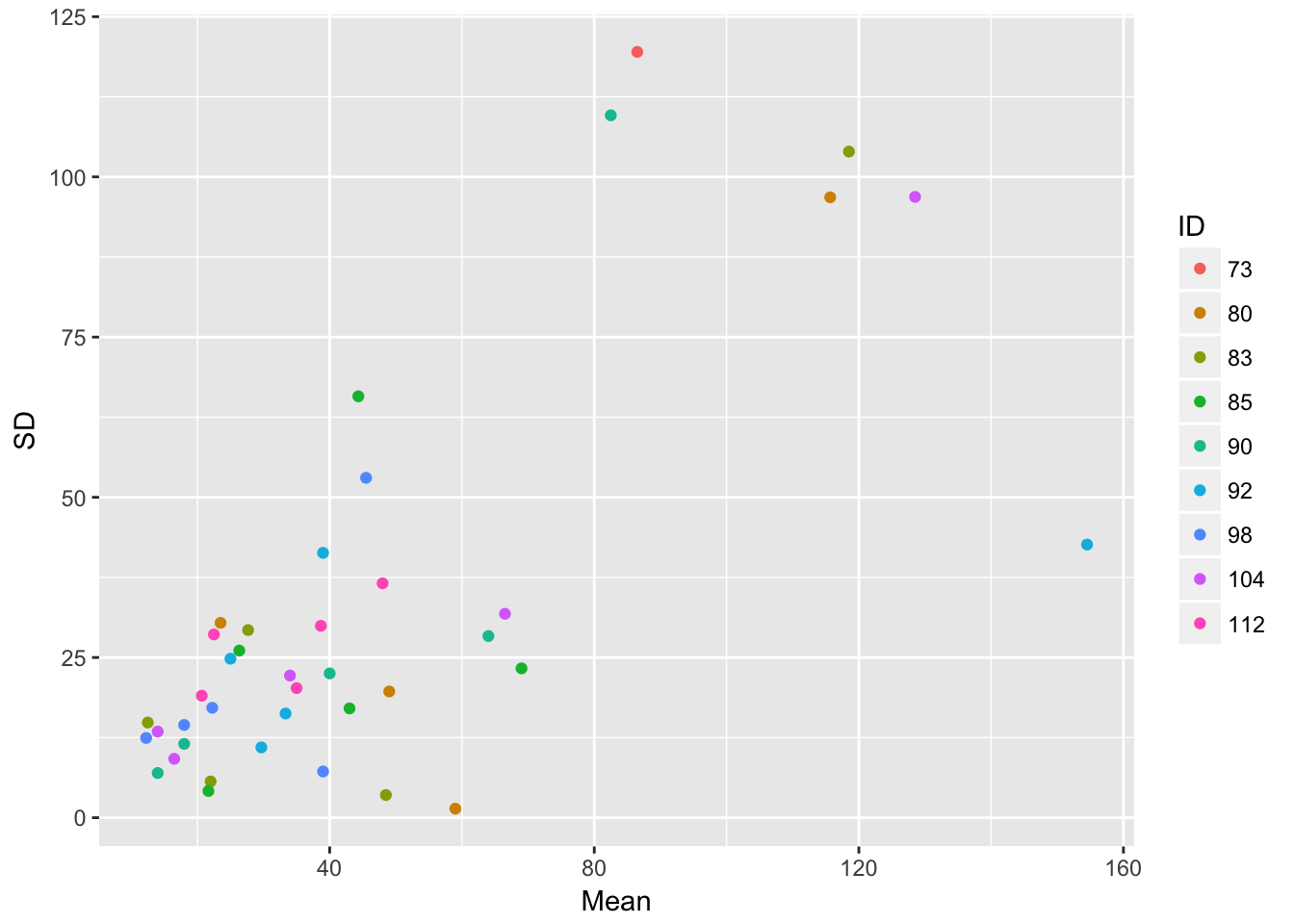
ggplot(aes(Mean, Var), data = sil_stats) + geom_point() + geom_smooth(method = "lm")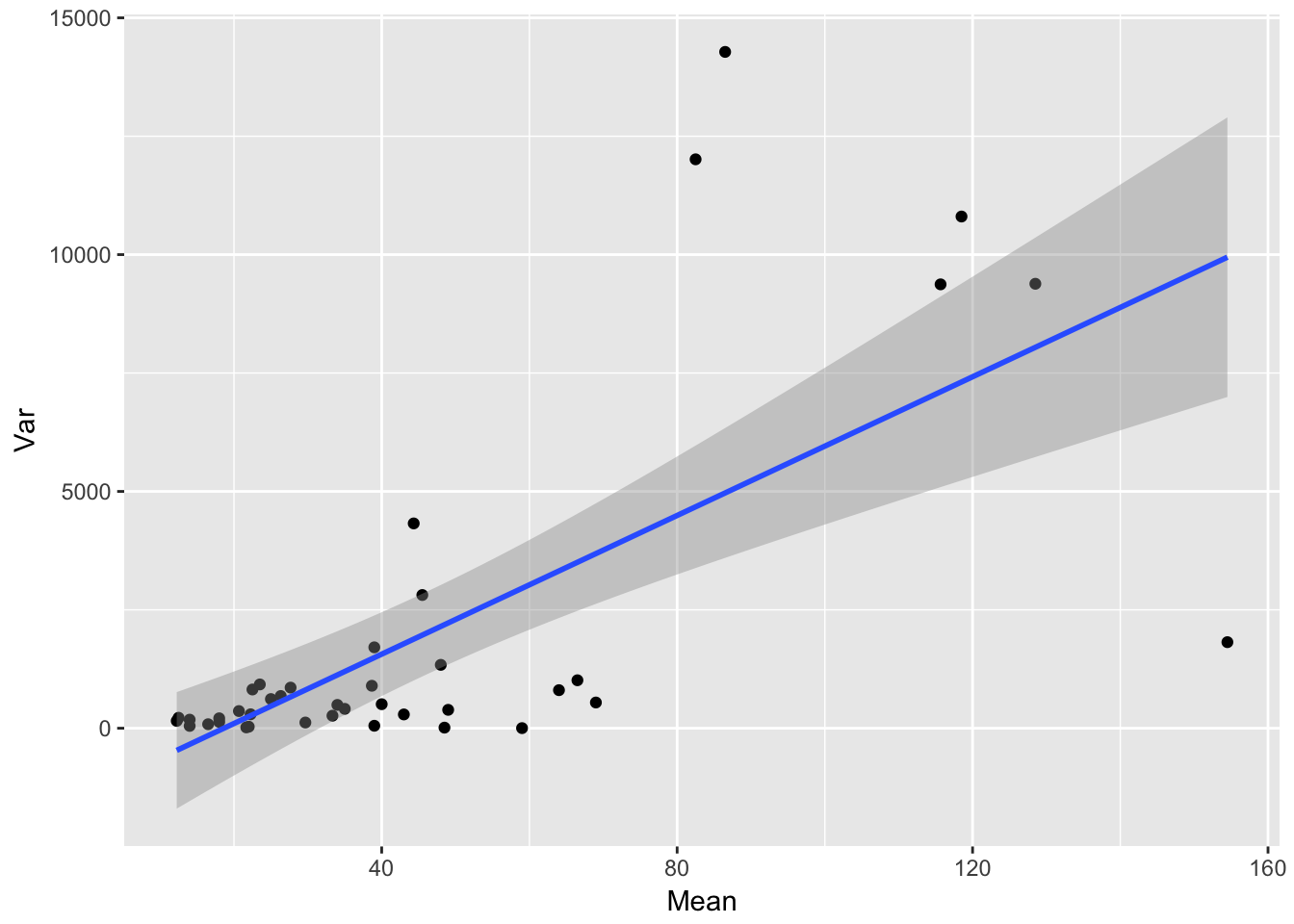
ggplot(aes(Mean, SD), data = sil_stats) + geom_point() + geom_smooth(method = "lm")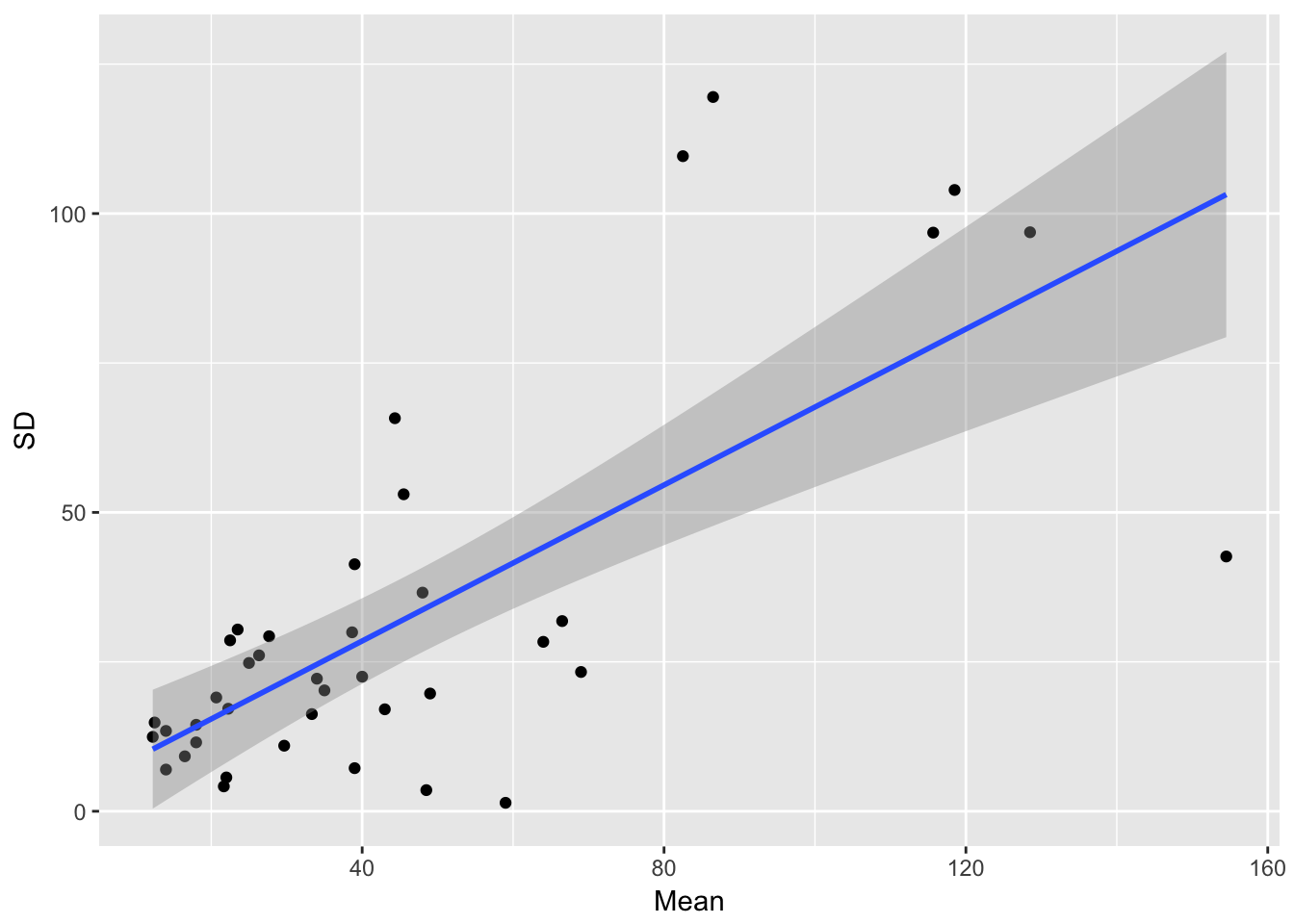 So, despite the lack of relationship between siliques and seedlings, we do see that seedling production is structured by runway, and that the SD scales roughly linearly with the mean (althoug not in a way that bives a constant CV)
So, despite the lack of relationship between siliques and seedlings, we do see that seedling production is structured by runway, and that the SD scales roughly linearly with the mean (althoug not in a way that bives a constant CV)
We can also look at population-level structure in the density-dependent populations.
seed_data <- group_by(popLer, ID, Pot) %>%
mutate(Nm1 = lag(Seedlings))
#seed_data$Nm1 <- lag(popLer$Seedlings)
seed_data <- subset(seed_data, Treatment == "C" & Generation > 1 & Gap == "1p")
seed_data$GenID <- with(seed_data, interaction(Gen, ID))
anova(lm(log(Seedlings/Nm1) ~ log(Nm1) + Gen + GenID, data = seed_data)) Analysis of Variance Table
Response: log(Seedlings/Nm1)
Df Sum Sq Mean Sq F value Pr(>F)
log(Nm1) 1 359.27 359.27 750.8986 < 2.2e-16 ***
Gen 4 38.89 9.72 20.3221 1.105e-12 ***
GenID 45 33.35 0.74 1.5489 0.0328 *
Residuals 115 55.02 0.48
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1ggplot(aes(log(Nm1), log(Seedlings/Nm1), group = GenID, color = Gen), data = seed_data) +
geom_point() + geom_smooth(method = "lm", se = FALSE)Warning: Removed 36 rows containing non-finite values (stat_smooth).Warning: Removed 36 rows containing missing values (geom_point).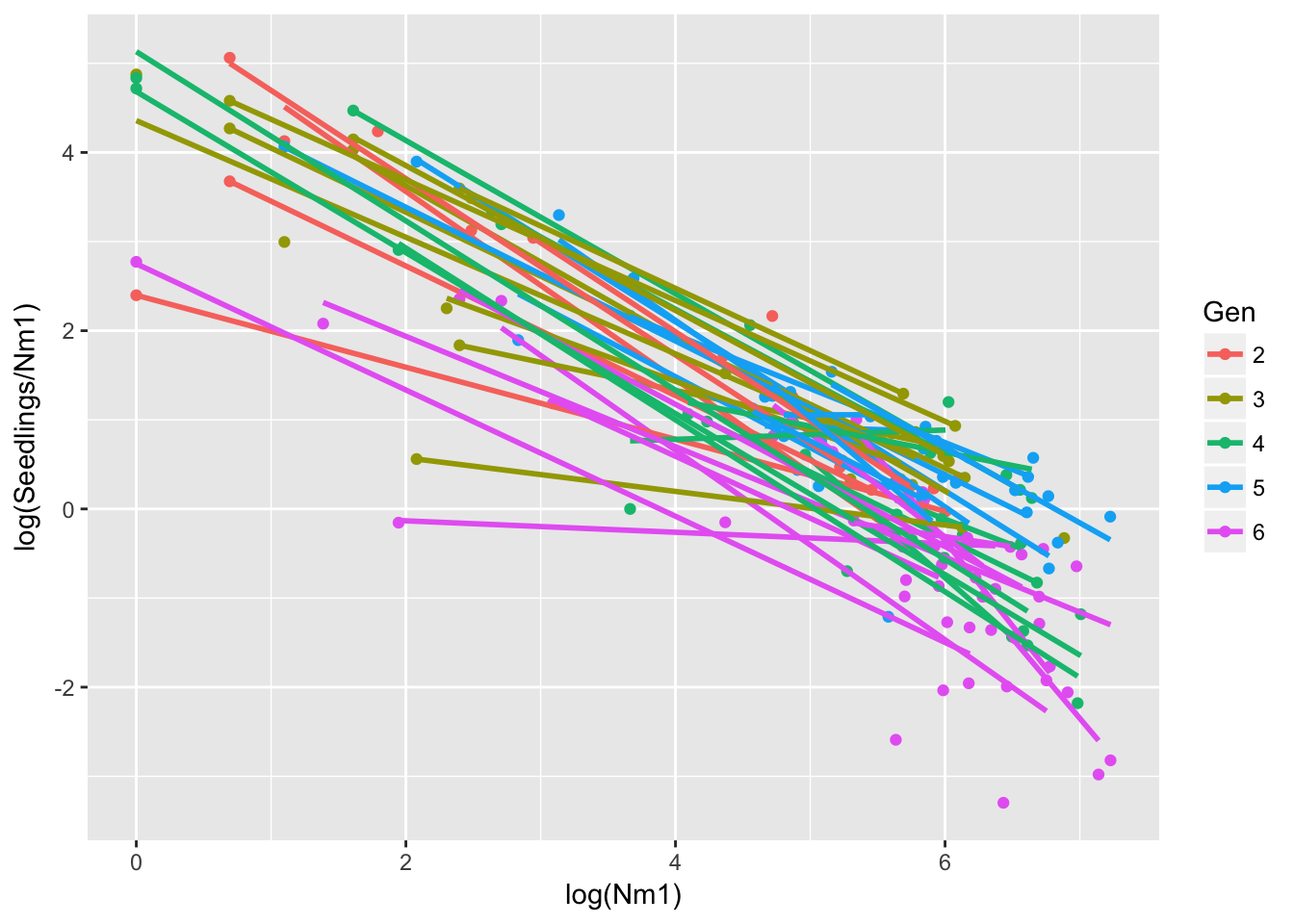 So the population-level variation is still here.
So the population-level variation is still here.